



氨基糖苷类抗生素的研究进展
姜智腾,李娜,李嘉钰,刘文婷,刘洪亮*
作者单位:山东良福制药有限公司
*E-mail:hongliang.liu liangfu.com.cn
【摘要】
氨基糖苷类抗生素是一种有效的广谱抗生素,主要用于治疗革兰氏阴性细菌感染。近年来,日益严重的细菌耐药问题引起社会各界的广泛关注,越来越多的科研工作者对氨基糖苷类抗生素重新产生兴趣,期望寻找到更好的解决办法。本文结合近年来氨基糖苷类抗生素类抗生素相关文献,介绍了氨基糖苷类抗生素在克服耐药或改变其生物活性方面进行的化学结构修饰。
关键词:氨基糖苷类抗生素,耐药性,抗菌活性,结构修饰
【前言】
抗生素是由某些微生物产生的、能抑制微生物和其他细胞增殖的物质,它的发现对人类健康产生了深远的影响。目前较常用的抗生素是β-内酰胺类、大环内酯类、喹诺酮类、四环素类和氨基糖苷类。而在这些抗生素中,氨基糖苷类因其广谱活性,具有快速抑菌和杀菌作用而受到特别关注[1]。20世纪60年代到70年代,氨基糖苷类药物在临床上得到了广泛的应用。然而,近年来由于毒副作用(耳毒性、肾毒性、神经肌肉阻断等)以及受到头孢类和喹诺酮类抗生素广泛应用的影响,其应用受到了相应的限制。但因为氨基糖苷类抗生素对铜绿假单胞菌、肺炎杆菌、大肠杆菌等常见革兰阴性杆菌的抗生素后效应较长、抗菌谱广泛,仍然是治疗革兰阴性杆菌和结核杆菌感染的一类重要药物[2-5]。
1、氨基糖苷类抗生素的发展
氨基糖苷类抗生素是一类由放线菌和细菌产生的代谢产物,其中一些已经在临床上应用了几十年。人类历史上第一个氨基糖苷类抗生素是链霉素,是在1944年从链霉菌分泌物中分离获得的[6]。随后,人们在1948年从弗氏链霉菌中分离出来新霉素[7]。1957年,从链霉菌中提取出卡那霉素[8]。1963年,人们从小单孢菌发酵液中成功分离出了庆大霉素[9]。这些天然抗生素主要用于治疗革兰氏阴性菌感染(图1)。随着抗生素在临床上的广泛应用,相应的耐药性问题也随之而来[10-11]。
为了克服这种对天然抗生素的耐药性,从20世纪70年代开始相继开发出了一批半合成氨基糖苷类抗生素,如阿米卡星(1972年)、奈替米星(1989年)、异帕霉素(1978年)、阿贝卡星(1995年)(图1)。但随着氨基糖苷类抗生素数量日益增多,微生物产生的耐药性也日益增强,这已成为影响人类健康的主要问题之一。因此,我们需要进一步研究设计新型且更有效的抗生素,来应对日益严峻的耐药性问题。
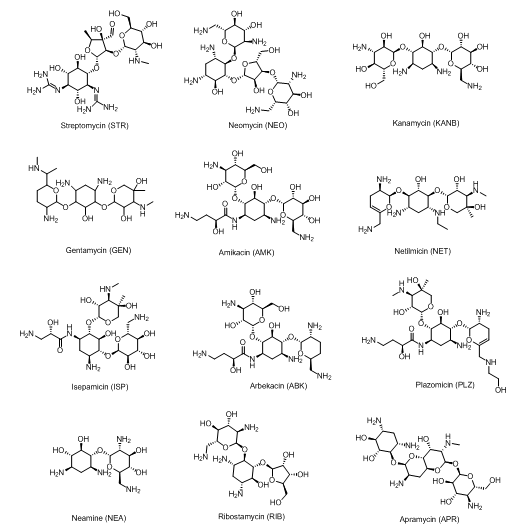
图1 一些天然氨基糖苷类抗生素和半合成氨基糖苷类抗生素
2、氨基糖苷类抗生素的耐药机制
氨基糖苷类抗生素通过与细胞30S核糖体亚单位的16S rRNA解码区的A部位相结合,继而诱导mRNA错误编码并合成错误蛋白,阻断mRNA和tRNA易位,从而抑制核糖体再循环[12-16]。核糖体再循环是在蛋白质合成终止后进行的,抑制核糖体再循环可导致细菌死亡[17]。
经大量研究发现,细菌对氨基糖苷类抗生素产生耐药性主要是通过以下几种方式:1)核糖体突变[18-20];2)16S rRNA甲基化酶修饰核糖体[21];3)氨基糖苷修饰酶[22];4)细胞膜转运减少或外膜通透性改变[23];5)主动外排系统降低细胞内氨基糖苷浓度[24]。其中,氨基糖苷修饰酶(AMEs)通过对氨基糖苷进行结构修饰,催化乙酰基、核苷酸基或磷酸基团转移到氨基环醇或糖苷的-OH或-NH2位置(图2),是迄今为止对这些抗生素产生耐药性的最重要的临床机制[25]。
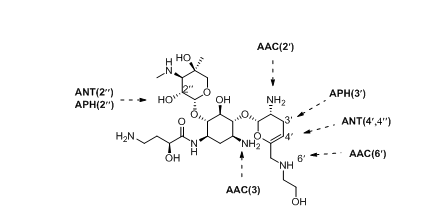
图2 氨基糖苷修饰酶(AMEs)在普拉佐米星的作用位点
3、氨基糖苷类抗生素的结构修饰
早在20世纪70年代,人们已经开始对氨基糖苷进行化学结构修饰,开发出一系列半合成氨基糖苷类抗生素,如奈替米星、异帕霉素、普拉佐米星[26]。
为了克服细菌病原体的耐药性及临床毒性,开发制备新的分子并不是一件容易的事。然而,近年来选择性修饰氨基糖苷不同位置的合成策略取得了成功。本文介绍了氨基糖苷类抗生素克服耐药性或改变其生物活性方面的主要化学修饰方向。
3.1氨基糖苷类抗生素常用的结构修饰策略
在氨基糖苷类抗生素结构中最常被修饰的位置是伯胺或羟基(图3)[27]。在NEO的5''-羟基以及AMK、ABK、KANA、KANB和TOB的6''-羟基位置引入一个良好的离去基团(如三异丙基苯磺酰(TIBS))。引入TIBS片段需要两步进行,首先用叔丁氧羰基(Boc)或苄氧羰基(Cbz)保护所有氨基,然后在伯醇位置上引入TIBS。最后,TIBS基团在亲核试剂(例如,胺、重氮化物和硫醇)的作用下离去,生成中间体或最终产物。
PAR的5''位结构修饰策略与上述非常相似,唯一的区别在于,在5''位引入TIBS基团之前,需要先对4'和6'-羟基进行保护,一般使用缩醛与PhCH(OMe)2。此外,另一种经常使用的结构修饰策略是通过碱催化氨基糖苷类抗生素与N-羟基琥珀酰亚胺酯反应,从而在6'-、6'''-或6'-、4'''-NH位置上引入羰基。
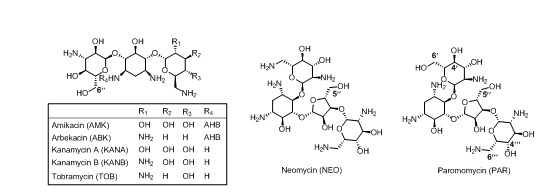
图3 氨基糖苷类抗生素的结构修饰位点
3.2氨基糖苷类抗生素的核心修饰策略
由于氨基糖苷类抗生素的氨基和羟基在与16S rRNA的A位点相互作用中起着关键作用,它们的修饰可能对氨基糖苷衍生物的活性产生很大影响。因此对氨基糖苷核心修饰主要集中在氨基和羟基基团上。(1)许多氨基糖苷的3位氨基碱度较低,可通过重氮化试剂一步将氨基转化为叠氮化物,并且具有较高的区域选择性[28];(2)羟基官能团转化为叠氮化物、胺或醛;(3)烯丙胺转化为α、β-不饱和醛(例如:维达霉素C2和C2a的全合成过程)[29];(4)利用Garegg-Samuelsson脱氧反应将α、β-不饱和醛合成3',4'-二脱氧氨基糖苷;(5)羟基基团被各种胺和卤素差向异构化或取代,可对铜绿假单胞菌和金黄色葡萄球菌具有良好的抑菌活性[30]。
3.3氨基糖苷类抗生素的烷基化和芳基化合成
在氨基糖苷核心上引入烷基或芳基来生成两亲性氨基糖苷是一种常见的修饰方法。通过把氨基糖苷的阳离子性质和烷基的疏水性质结合,得到的两亲体便可以通过疏水、静电等方式与细菌膜相互作用。氨基糖苷与烷基或芳基分子可以通过形成硫醚、醚、胺、三唑、酰胺以及氨基甲酸酯键来构建两亲性氨基糖苷。这种策略具有提供能够选择性靶向细菌和真菌膜的氨基糖苷、提高活性以及避免在氨基糖苷修饰酶作用下失活的优点[27]。
相关研究小组已经报道了两亲性新霉素的合成和抗菌研究,大部分的两亲性新霉素对氨基糖苷耐药菌具有广谱性和较高的生物活性。例如,带有线性酰基的5"-氨基霉素对耐甲氧西林金黄色葡萄球菌(MRSA)和耐万古霉素肠球菌(VRE)显示出突出的抗菌活性[31]。
3.4氨基糖苷类抗生素的酰基化合成
经研究发现,N-酰化的氨基糖苷在增加抗菌活性的同时,也可以抑制氨基糖苷修饰酶作用。单-N-酰化的反应利用了N-羟基琥珀酰亚胺酯偶联,通过碱催化与相应的N-羟基琥珀酰亚胺酯进行反应[27,32]。相关实验表明,特定氨基糖苷和酰基的组合对改善N-酰化类似物的生物活性具有较大的影响[33]。另一种合成N-酰化氨基糖苷的方法是通过氨基糖苷的氨基与多种羧酸选择性偶联实现的。例如:ABK的4"-和6"-N-酰基衍生物是通过ABK与多种脂肪族和芳香族羧酸的偶联而合成[34]。ABK首先选择性的对6"-羟基磺化,接着引入叠氮化物、胺化、与羧酸进行N-酰化反应。接着在改造后的6"-N-酰化衍生物结构上引入4-氨基-2-羟基丁酰胺片段(AHB),显示出对金黄色葡萄球菌的有效抗菌活性。
最后一种方法是对氨基糖苷阿帕霉素、卡那霉素和新霉素的多种N-芳基/酰化O-磺化类似物进行合成[27]。O-磺化N-芳酰基氨基糖苷的合成分两步完成,APR、KANA和NEO首先在碱性条件下与相应的N-酰基琥珀酰亚胺酯或酰氯偶联,转化为相应的N-酰基衍生物,然后用吡啶三氧化硫络合物磺化。实验结果表明,AGs和N-芳基/酰基片段的结构对不同的局部异构酶具有不同的选择性抑制作用[35]。
3.5氨基糖苷与生物活性分子的偶联
在前面我们已经了解了在氨基糖苷结构上引入烷基、芳基等基团或者将氨基或羟基转化为其他结构对细菌耐药性的影响,接着将介绍氨基糖苷与生物活性分子的偶联。根据偶联组分的不同,将其分为四类:1)利用乙酰辅酶A与氨基糖类抗生素生成一种双底物抑制剂,对6'-氨基位置上无保护基的氨基糖苷(如:NEA、RIB等)进行区域和化学选择性衍生化。研究发现,具有末端羟基基团但不具有N-乙酰转移酶抑制活性的NEA-泛硫醇类似物可作为前药,并利用细胞的辅酶A的生物合成途径产生具有活性的N-乙酰转移酶抑制剂[36]。2)在临床上相关抗生素在与其16S rRNA靶标结合不相关的位置与氨基糖苷共价连接,可得到氨基糖苷-药物偶联物[37-38]。一种头孢菌素(类头孢菌素)在C-7位点上与氟喹诺酮类药物通过羧基键连接而成,但由于毒副作用限制了其临床应用。然而,通过将头孢菌素的细胞壁合成抑制作用与喹诺酮类药物的DNA旋切抑制活性结合在一起,使得该类衍生物(如:去乙酰头孢噻肟与氟罗沙星的偶联药物)对肠杆菌科、葡萄球菌、斯嗜海杆菌等具有强效活性[38-39]。3)氨基糖苷与其他生物相关的小分子(如:碱基、聚乙二醇、荧光分子等)通过硫脲键、三唑键和亲核取代反应形成氨基糖苷-小分子共轭物[27]。该方法是为了提高抗生素对RNA的结合特异性以及增强抗菌活性。4)氨基糖苷与氨基酸或某些肽序列相结,既可以增强对RNA靶点的亲和力,也可以增强抗菌活性[40]。已经证明l-精氨酸与卡那霉素和庆大霉素的偶联物能够通过HIV-I RNA 5位末端的特定核苷酸序列与HIV-I转激活响应(TAR)部分结合,这是病毒启动转激活和复制所必需的[41]。TAR与反激活子转录(Tat)蛋白的相互作用,对病毒复制起着较大的影响。因此,开发与TAR结合并干扰这些相互作用的化合物是今后氨基糖苷类抗生素研究的一个方向[26]。
【结论】
近年来,由于临床上出现了越来越多的耐药菌株,氨基糖苷类抗生素作为临床使用的重要抗菌药物再度成为药物研究关注的重点。本文主要对氨基糖苷类抗生素在克服耐药或改变其生物活性方面进行的化学结构修饰进行了介绍。氨基糖苷结构上具有反应活性的氨基与羟基的转化及这类抗生素的其他类型生物活性的修饰,为提高抗生素对耐药菌株活性提供了研究方向。我们相信,随着科研工作者们对氨基糖苷类抗生素的不断深入研究,将会开发出更多的具有高效生物活性的药物。
参考文献:
[1]Xie J,Talaska A E,Schacht J.New developments in aminoglycoside therapy and ototoxicity[J].Hear.Res.,2011,281:28-37.
[2]Krause K M,Serio A W,Kane T R,et al.Aminoglycosides:an overview[J].Cold Spring Harbor Perspect.Med.,2016,6,a027029.
[3]Ramirez M S,Tolmasky M E.Aminoglycoside modifying enzymes[J].Drug Resistance Updates,2010,13(6):151-171.
[4]Serio A W,Keepers T,Andrews L,et al.Aminoglycoside revival:review of a historically important class of antimicrobials undergoing rejuvenation[J].EcoSal Plus,2018,8(1),DOI:10.1128/ecosalplus.ESP-0002-2018.
[5]Thy M,Timsit J,Montmollin E de.Aminoglycosides for the treatment of severe infection due to resistant gram-negative pathogens[J].Antibiotics,2023,12(5):860.
[6]Waksman S A.Streptomycin:background,isolation,properties,and utilization[J].Science,1953,118(3062):259-266.
[7]Waksman S A,Lechevalier H A.Neomycin,a new antibiotic active against streptomycin-resistant bacteria,including tuberculosis organisms[J].Science,1949,109(2830):305-307.
[8]Umezawa H,Ueda M,Maeda K,et al.,Production and isolation of a new antibiotic:kanamycin[J].J.Antibiot(Tokyo),1957,10(5):181-188.
[9]Weinstein M J,Luedemann G M,Oden E M,et al.,Gentamicin,1a new antibiotic complex from micromonospora[J].Journal of Medicinal Chemistry 1963,6(4):463-464.
[10]Fernández L,Breidenstein E B M,Hancock R E W.Creeping baselines and adaptive resistance to antibiotics[J].Drug Resist.Updat.,2011,14(1):1-21.
[11]Blair J M,Webber M A,Baylay A J,et al.Molecular mechanisms of antibiotic resistance[J].Nat.Rev.Microbiol.,2015,13(1):42-51.
[12]Visal C S,Prasobh G R,Sheeja R A G,et al.A review on aminoglycoside antibiotics[J].IAJPS,2020,7(10):5-9.
[13]Magnet S,Blanchard,J S.Molecular insights into aminoglycoside action and resistance[J].Chem.Rev.,2005,105(2):477-497.
[14]Feldman M B,Terry D S,Altman R B,et al.Aminoglycoside activity observed on single pre-translocation ribosome complexes[J].Nat.Chem.Biol.,2010,6(1):54-62.
[15]Kumar C G,Himabindu M,Jetty A.Microbial biosynthesis and applications of gentamicin:A critical appraisal[J].Crit.Rev.Biotechnol.,2008,28(3):173-212.
[16]Bottger E C,Crich D.Aminoglycosides:time for the resurrection of a neglected class of antibacterials?[J].ACS Infect.Dis.,2020,6:168-172.
[17]Houghton J L,Green K D,Chen W,et al.The future of aminoglycosides:The end or renaissance[J].Chem.Bio.Chem.,2010,11(7):880-902.
[18]Santos L C,Farmácia D de,Objetivo F,et al.Review:the molecular basis of resistance in mycobaterium tuberculosis[J].Open J.Med.Microbiol.,2012,2(1):24-36.
[19]Maus C E,Plikaytis B B,Shinnick T M.Molecular analysis of cross-resistance to capreomycin,kanamycin,amikacin,and viomycin in mycobacterium tuberculosis[J].Agents Chemother.,2005,49(8):3192-3197.
[20]Nessar R,Cambau E,Reyrat J M,et al.Mycobacterium abscessus:a new antibiotic nightmare[J].J.Antimicrob.Chemother.,2012,67(4):810-818.
[21]Gutierrez B,Douthwaite S,Gonzalezzorn B.Indigenous and acquired modifications in the aminoglycoside binding sites of Pseudomonas aeruginosa rRNAs[J].Rna Biol,2013,10(8):1324-1332.
[22]Ramirez M S,Tolmasky M E.Aminoglycoside modifying enzymes[J].Drug Resist.Updates,2010,13(6):151-171.
[23]Li X Z,Plésiat P,Nikaido H.The Challenge of efflux mediated antibiotic resistance in Gram-negative bacteria[J].CMR,2015,28(2):337-418.
[24]Li X Z,Nikaido H.Efflux-mediated drug resistance in bacteria:an update[J].Drugs,2009,69(12):1555-1623.
[25]Magaña A J,Sklenicka J,Pinilla C,et al.Restoring susceptibility to aminoglycosides:identifying small molecule inhibitors of enzymatic inactivation[J].RSC Med.Chem.,2023,14:1591-1602.
[26]Tevyashova1 A N,Shapovalova1 K S.Search for new drugs potential for the development of a new generation of aminoglycoside antibiotics[J].Pharmaceutical Chemistry Journal,2021,55(9):7-23.
[27]Chandrika N T,Garneau-T S.Comprehensive review of chemical strategies for the preparation of new aminoglycosides and their biological activities[J].Chem.Soc.Rev.,2018,47:1189-1249.
[28]Bastian A A,Warszawik E M,Panduru P,et al.Regioselective diazo-transfer reaction at the C3-position of the 2-desoxystreptamine ring of neamine antibiotics[J].Chemistry,2013,19:9151-9154.
[29]Hanessian S,Szychowski J,Maianti J P.Synthesis and comparative antibacterial activity of verdamicin C2 and C2a.A new oxidation of primary allylic azides in dihydro[2H]pyrans[J].Org.Lett.,2009,11(2):429-432.
[30]Hiraiwa Y,Usui T,Akiyama Y,et al.Synthesis and antibacterial activity of 5-deoxy-5-episubstituted arbekacin derivatives[J].Med.Chem.Lett.,2007,17(13):3540-3543.
[31]Takemoto J Y,Altenberg G A,Poudyal N,et al.Amphiphilic aminoglycosides:modifications that revive old natural product antibiotics[J].Front.Microbiol.,2022,13,doi:10.3389/fmicb.2022.1000199.
[32]Shaul P,Green K D,Rutenberg R,et al.Assessment of 6′-and 6′′′-N-acylation of aminoglycosides as a strategy to overcome bacterial resistance[J].Org.Biomol.Chem.,2011,9(11):4057-4063.
[33]Chandrika N T,Green K D,Houghton J L,et al.Synthesis and biological activity of mono-and di-N-acylated aminoglycosides[J].ACS Med.Chem.Lett.,2015,6(11):1134-1139.
[34]Sasaki K,Kobayashi Y,Kurihara T,et al.Synthesis and antibacterial activity of 4″or 6″-alkanoylamino derivatives of arbekacin[J].J.Antibiot.,2015,68:741-747.
[35]Fenner A M,Oppegard L M,Hiasa H,et al.Selective inhibition of bacterial and human topoisomerases by N arylacyl O sulfonated aminoglycoside derivatives[J].ACS Med.Chem.Lett.,2013,4(5):470-474.
[36]Vong K,Tam I S,Yan X,et al.Inhibitors of aminoglycoside resistance activated in cells[J].ACS Chem.Biol.,2012,7(3):470-475.
[37]Zhi C X,Long Z Y,Manikowski A,et al.Hybrid antibacterials.DNA polymerase:topoisomerase inhibitors[J].J.Med.Chem.,2006,49(4):1455-1465.
[38]Pokrovskava V,Belakhov V,Hainrichson M,et al.Design,Synthesis,and Evaluation of Novel Fluoroquinolone-Aminoglycoside Hybrid Antibiotics[J].J.Med.Chem.,2009,52(8):2243-2254.
[39]Pokrovskaya V,Belakhov V,Hainrichson M,et al.Design,synthesis,and evaluation of novel fluoroquinolone-aminoglycoside hybrid antibiotics[J].J.Med.Chem.,2009,52:2243-2254.
[40]Aradi K,Giorgio A Di,Duca M.Aminoglycoside conjugation for RNA targeting:antimicrobials and beyond[J].Chem.Eur.J.,2020,26(54):12273-12309.
[41]Hermann T.Small molecules targeting viral RNA[J].Wiley Interdiscip.Rev.RNA,2016,7(6):726-743.



